Gestación de una madre sana de 31 años. Embarazo controlado. A las 38+5 semanas de gestación nace recién nacido mediante parto instrumentado por bradicardia fetal mantenida. Serologías maternas: inmune a Rubeola y Toxoplasma, Lúes, VHB, VHC, VIH, Zika, Herpes y Parvovirus negativas. PCR-CMV negativo. Cultivo recto-vaginal para SGB negativo. Nace con una prueba de Apgar al minuto de 6 y a los 5 minutos de 8; pHau: 7.30. Ecografías prenatales: a las 36 semanas se detecta retraso de crecimiento intrauterino, calcificaciones parenquimatosas encefálicas, dilatación ventricular leve tetracameral, y calcificaciones intestinales sugestivas de infección connatal. A las 38+5 semanas progresa a CIR grave, microcefalia, ventriculomegalia, ecogenicidad periventricular marcada, un cuerpo calloso adelgazado, y calcificaciones parenquimatosas diseminadas. En la exploración física al nacer destaca, un peso de nacimiento: 2384g (p2), longitud: 46,5cm (p5) y perímetro cefálico: 29,5cm (<p1). Microcefalia con frente huidiza. En el examen neurológico destaca una hipotonía generalizada.
- Infección connatal (TORCH).
- Síndrome Pseudo-TORCH.
- Síndrome Aicardi-Goutieres.
- Todas las respuestas anteriores pueden ser ciertas.
Respuesta correcta : Todas las respuestas anteriores pueden ser ciertas.
En el corte coronal y parasagital de la ecografía cerebral (Fig. 1A y 1B) se aprecian calcificaciones parenquimatosas, en ganglios de la base y en el tálamo. También se aprecia un quiste de septum prominente para la edad gestacional a término.
En el corte axial de la RM cerebral (Fig. 1C y 1D) se aprecia un aumento de señal de sustancia blanca con una ventriculomegalia y un quiste de septum enorme.
La asociación de retraso de crecimiento intrauterino, la microcefalia y las calcificaciones intracraneales son datos clínicos característicos del síndrome TORCH causado por infecciones intrauterinas, pero pueden estar también determinados por entidades genéticas autosómicas recesivas que remedan el cuadro de presentación clínica de las infecciones connatales, y que son conocidas como síndrome pseudo-TORCH. Existen varios tipos:
El síndrome pseudo TORCH-1, es debido a la mutación en el gen de la ocludina (OCLN) localizado en el cromosoma 5q13. Los individuos afectados tienen microcefalia congénita, calcificaciones intracraneales, giro simplificado y polimicrogiria y retraso grave del desarrollo.
El síndrome pseudo TORCH-2, es causado por una mutación homocigótica o heterocigótica compuesta en el gen USP18 en el cromosoma 22q11. Se caracteriza por la aparición prenatal de hemorragia intracraneal, calcificación, malformaciones cerebrales, disfunción hepática y, a menudo, trombocitopenia. Los individuos afectados tienden a padecer insuficiencia respiratoria y convulsiones, y mueren en la infancia.
El síndrome pseudo TORCH-3, es causado por una mutación homocigótica en el gen STAT2 en el cromosoma 12q13. Muestran retraso en el desarrollo con episodios agudos de fiebre y afectación de órganos multisistémicos, que incluyen coagulopatía, elevación de las enzimas hepáticas y proteinuria, a menudo asociadas con microangiopatía trombótica. Las imágenes cerebrales muestran calcificaciones intracraneales progresivas, anomalías de la sustancia blanca y, a veces, atrofia cerebral o cerebelosa.
En estas entidades pseudo-TORCH, la microcefalia y las calcificaciones cerebrales congénitas, junto con la clínica de encefalopatía son las principales características clínicas. Los hallazgos que recuerdan a las infecciones congénitas como la ictericia, la trombocitopenia y la hepatomegalia, afectan a menos del 50% de los pacientes.
La microcefalia y las calcificaciones intracraneales pueden ocurrir también en el síndrome de Aicardi-Goutiéres (SAG) y en las encefalopatías mitocondriales.
El SAG es una encefalopatía subaguda hereditaria caracterizada por calcificación progresiva de los ganglios de la base y leucodistrofia de la sustancia blanca con linfocitosis y aumento del INF-alfa en el LCR, mediado por el sistema inmune. La microcefalia no suele estar presente al nacimiento.
En nuestro paciente el diagnóstico etiopatogénico se orientó inicialmente al estudio de las infecciones connatales que fueron descartadas tanto intraútero como en estudios postnatales, por lo que se consideraron otros diagnósticos como las entidades genéticas incluidas bajo el paraguas del síndrome pseudo-TORCH. El estudio genético de este paciente está pendiente de resultado.
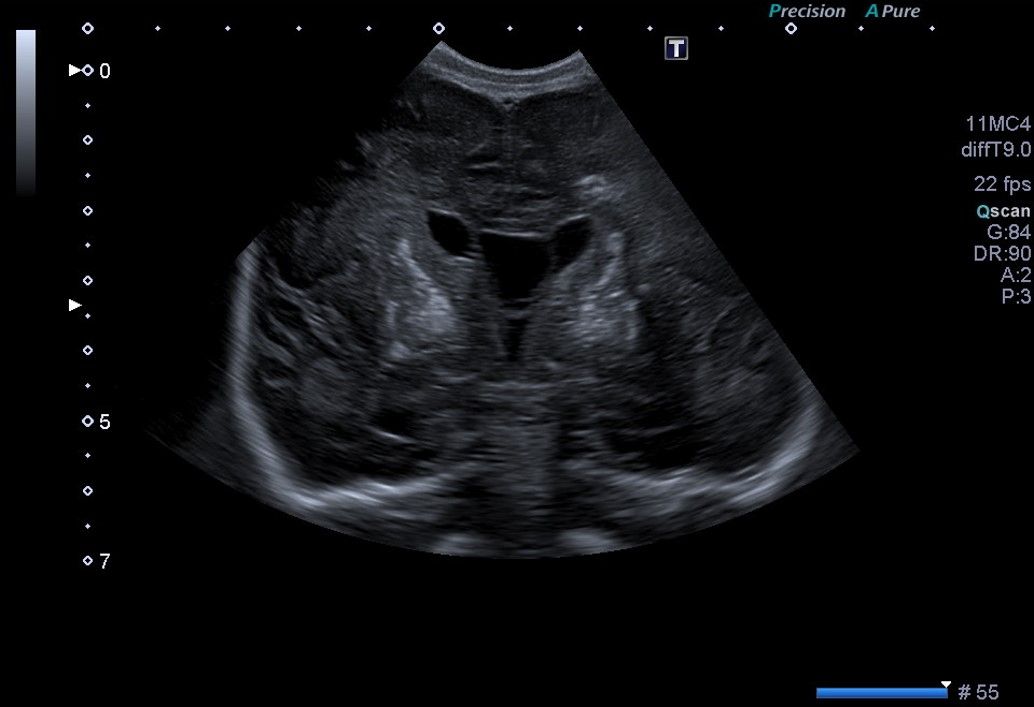
Figura 1A. USc. Corte coronal (C3).
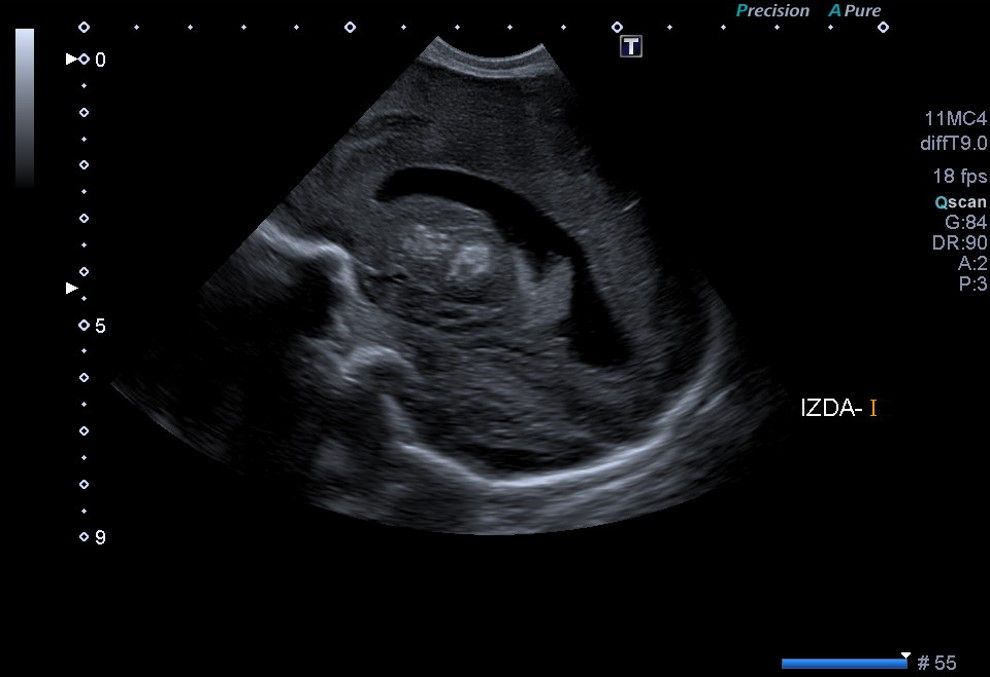
Figura 1B. USc. Corte parasagital izquierdo (S2)
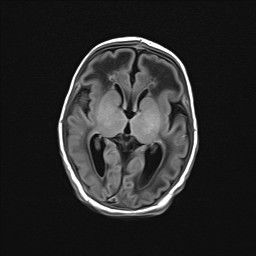
Figura 1C. RM cerebral. Corte axial en T1-WI.
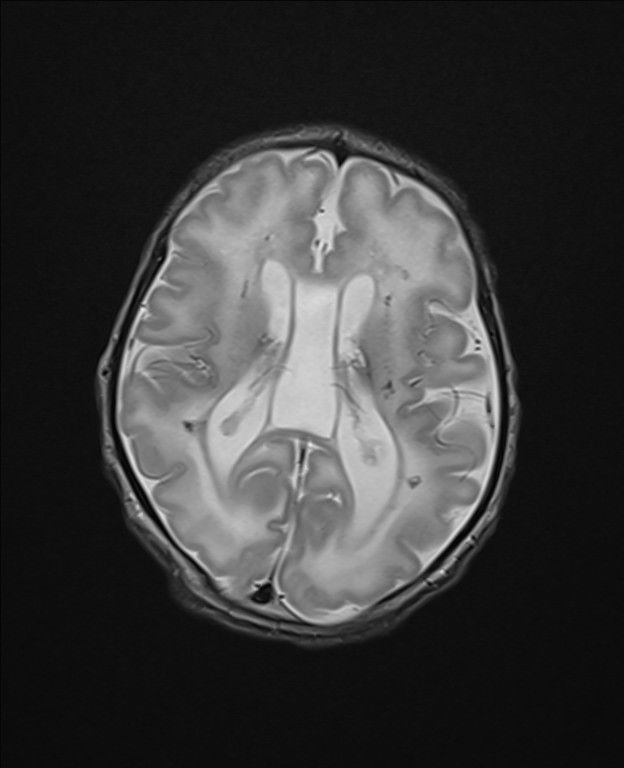
Figura 1D. RM cerebral. Corte axial en T2-WI.